Recipes¶
Various recipes implemented in Auto-FOX.
A set of functions for analyzing and plotting ARMC results. |
|
A set of functions for creating .psf files. |
|
A set of functions for analyzing ligands. |
|
A set of functions for calculating time-resolved distribution functions. |
|
Recipes for computing the similarity between trajectories. |
|
Recipe for creating GROMACS .top files from an .xyz and CHARMM .rtf and .str files. |
FOX.recipes.param¶
A set of functions for analyzing and plotting ARMC results.
Examples
A general overview of the functions within this module.
>>> import pandas as pd
>>> from FOX.recipes import get_best, overlay_descriptor, plot_descriptor
>>> hdf5_file: str = ...
>>> param: pd.Series = get_best(hdf5_file, name='param') # Extract the best parameters
>>> rdf: pd.DataFrame = get_best(hdf5_file, name='rdf') # Extract the matching RDF
# Compare the RDF to its reference RDF and plot
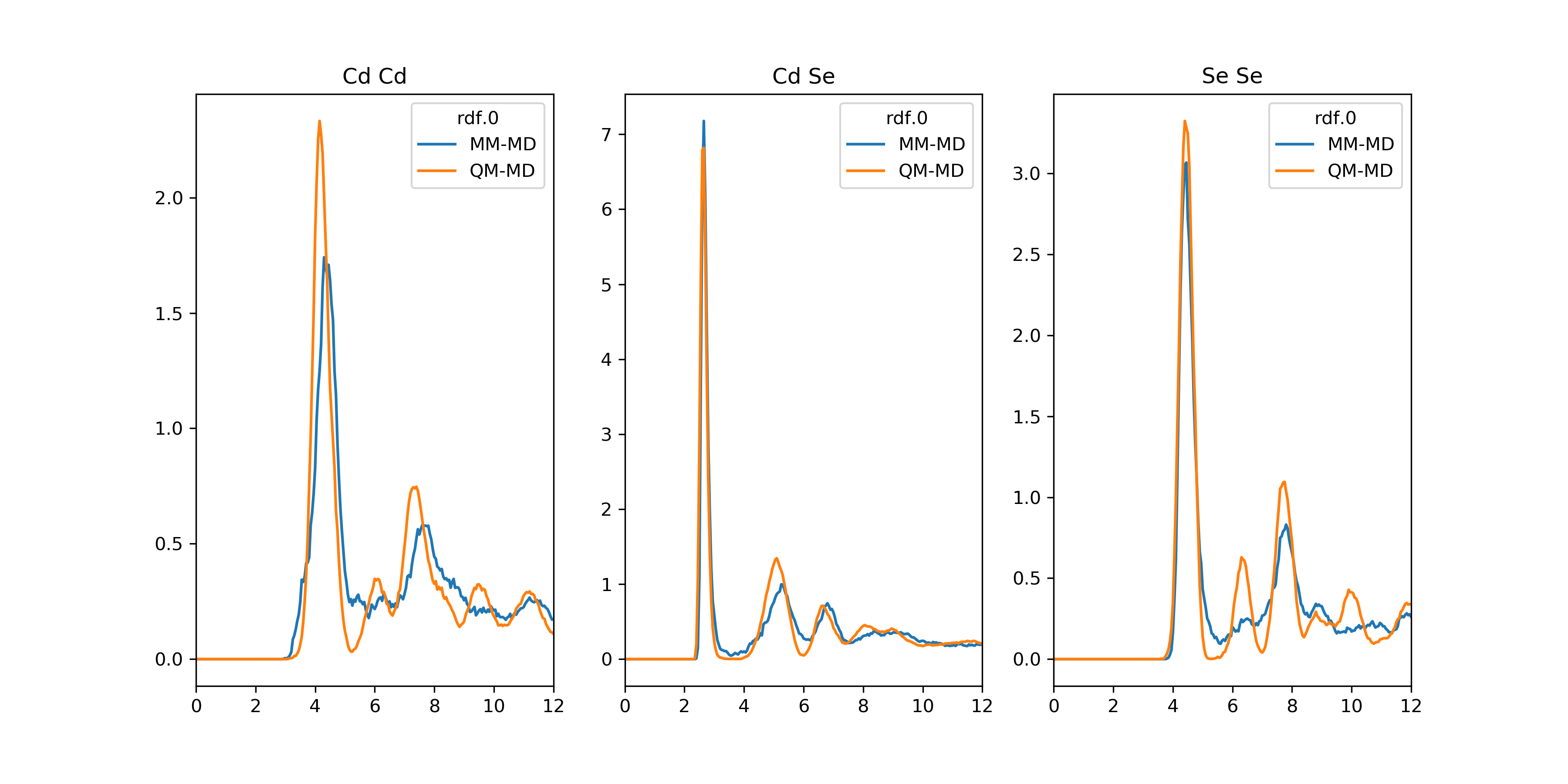
>>> rdf_dict = overlay_descriptor(hdf5_file, name='rdf')
>>> plot_descriptor(rdf_dict)
Examples
A small workflow for calculating for calculating free energies using distribution functions such as the radial distribution function (RDF).
>>> import pandas as pd
>>> from FOX import get_free_energy
>>> from FOX.recipes import get_best, overlay_descriptor, plot_descriptor
>>> hdf5_file: str = ...
>>> rdf: pd.DataFrame = get_best(hdf5_file, name='rdf')
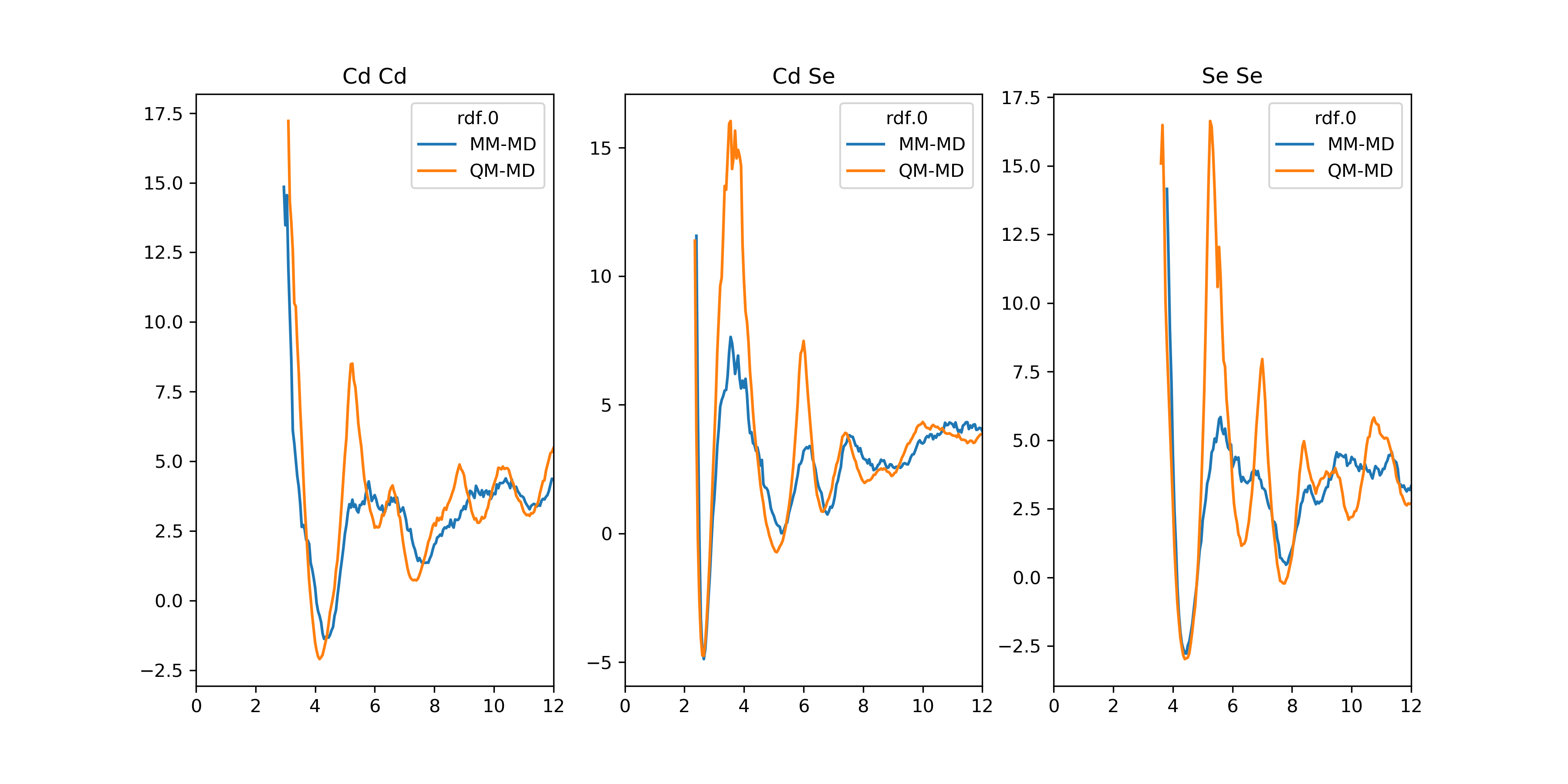
>>> G: pd.DataFrame = get_free_energy(rdf, unit='kcal/mol')
>>> rdf_dict = overlay_descriptor(hdf5_file, name='rdf)
>>> G_dict = {key: get_free_energy(value) for key, value in rdf_dict.items()}
>>> plot_descriptor(G_dict)
Examples
A workflow for plotting parameters as a function of ARMC iterations.
>>> import numpy as np
>>> import pandas as pd
>>> from FOX import from_hdf5
>>> from FOX.recipes import plot_descriptor
>>> hdf5_file: str = ...
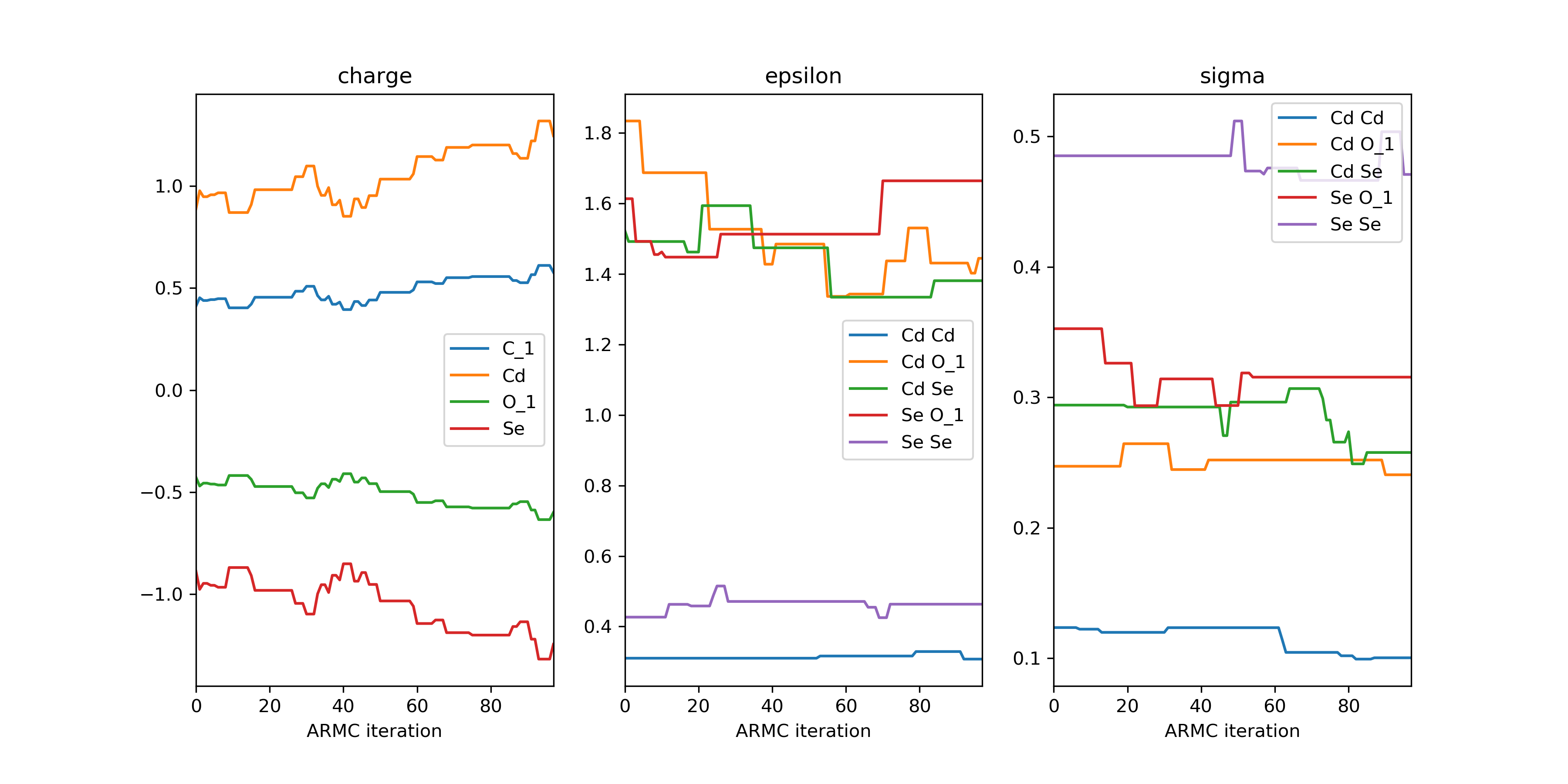
>>> param: pd.DataFrame = from_hdf5(hdf5_file, 'param')
>>> param.index.name = 'ARMC iteration'
>>> param_dict = {key: param[key] for key in param.columns.levels[0]}
>>> plot_descriptor(param_dict)
This approach can also be used for the plotting of other properties such as the auxiliary error.
>>> ...
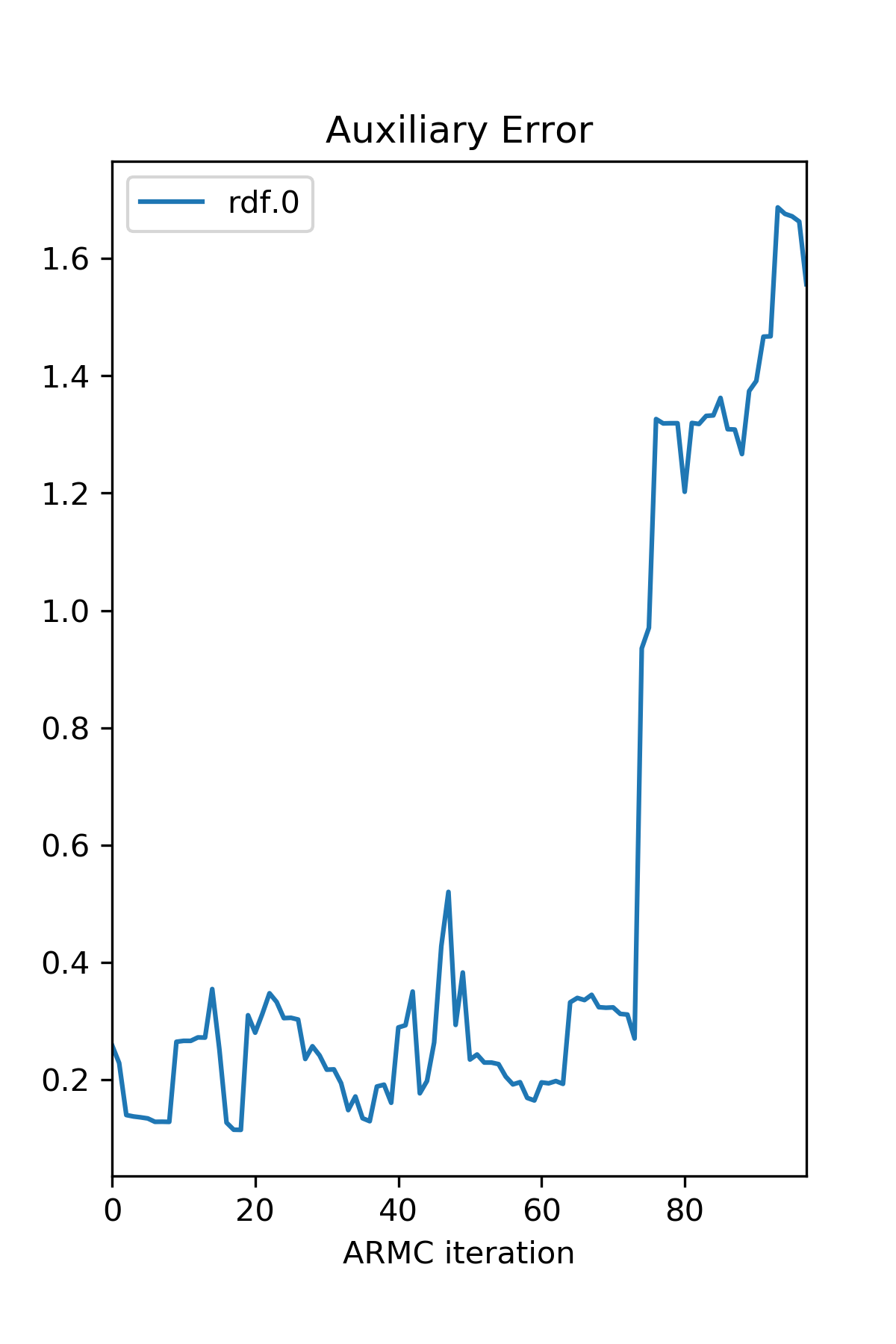
>>> err: pd.DataFrame = from_hdf5(hdf5_file, 'aux_error')
>>> err.index.name = 'ARMC iteration'
>>> err_dict = {'Auxiliary Error': err}
>>> plot_descriptor(err_dict)
On occasion it might be desirable to only print the error of, for example, accepted iterations.
Given a sequence of booleans (bool_seq), one can slice a DataFrame or Series (df) using
df.loc[bool_seq].
>>> ...
>>> acceptance: np.ndarray = from_hdf5(hdf5_file, 'acceptance') # Boolean array
>>> err_slice_dict = {key: df.loc[acceptance], value for key, df in err_dict.items()}
>>> plot_descriptor(err_slice_dict)
Index¶
|
Return the PES descriptor or ARMC property which yields the lowest error. |
|
Return the PES descriptor which yields the lowest error and overlay it with the reference PES descriptor. |
|
Plot a DataFrame or iterable consisting of one or more DataFrames. |
API¶
- FOX.recipes.get_best(hdf5_file, name, i=0, sum_error=None, err_dset='aux_error')[source]¶
Return the PES descriptor or ARMC property which yields the lowest error.
- Parameters:
hdf5_file (
str) – The path+filename of the ARMC .hdf5 file.name (
str) – The name of the PES descriptor, e.g."rdf". Alternatively one can supply an ARMC property such as"acceptance","param"or"aux_error".i (
int) – The index of the desired PES. Only relevant for PES-descriptors of state-averaged ARMCs.sum_error (
strorlist[str], optional) – Sum all the given aux errors for a given iteration when determining an optimum. IfNone, sum over all aux errors.err_dset (
str) – The name of the dataset containing the errors. Generally speaking one should pick either"aux_error"or"validation/aux_error".
- Returns:
A DataFrame of the optimal PES descriptor or other (user-specified) ARMC property.
- Return type:
pandas.DataFrameorpd.Series
- FOX.recipes.overlay_descriptor(hdf5_file, name='rdf', i=0, err_dset='aux_error')[source]¶
Return the PES descriptor which yields the lowest error and overlay it with the reference PES descriptor.
- Parameters:
hdf5_file (
str) – The path+filename of the ARMC .hdf5 file.name (
str) – The name of the PES descriptor, e.g."rdf".i (
int) – The index of desired PES. Only relevant for state-averaged ARMCs.err_dset (
str) – The name of the dataset containing the errors. Generally speaking one should pick either"aux_error"or"validation/aux_error".
- Returns:
A dictionary of DataFrames. Values consist of DataFrames with two keys:
"MM-MD"and"QM-MD". Atom pairs, such as"Cd Cd", are used as keys.- Return type:
- FOX.recipes.plot_descriptor(descriptor, show_fig=True, kind='line', sharex=True, sharey=False, **kwargs)[source]¶
Plot a DataFrame or iterable consisting of one or more DataFrames.
Requires the matplotlib package.
- Parameters:
descriptor (
pandas.DataFrameorIterable[pandas.DataFrame]) – A DataFrame or an iterable consisting of DataFrames.show_fig (
bool) – Whether to show the figure or not.kind (
str) – The plot kind to-be passed topandas.DataFrame.plot().sharex/sharey (
bool) – Whether or not the to-be created plots should share their x/y-axes.**kwargs (
Any) – Further keyword arguments for thepandas.DataFrame.plot()method.
- Returns:
A matplotlib Figure.
- Return type:
See also
get_best()Return the PES descriptor or ARMC property which yields the lowest error.
overlay_descriptor()Return the PES descriptor which yields the lowest error and overlay it with the reference PES descriptor.
FOX.recipes.psf¶
A set of functions for creating .psf files.
Examples
Example code for generating a .psf file.
Ligand atoms within the ligand .xyz file and the qd .xyz file should be in the exact same order.
For example, implicit hydrogen atoms added by the
from_smiles() functions are not guaranteed
to be ordered, even when using canonical SMILES strings.
>>> from scm.plams import Molecule, from_smiles
>>> from FOX import PSFContainer
>>> from FOX.recipes import generate_psf
# Accepts .xyz, .pdb, .mol or .mol2 files
>>> qd = Molecule(...)
>>> ligand: Molecule = Molecule(...)
>>> rtf_file : str = ...
>>> psf_file : str = ...
>>> psf: PSFContainer = generate_psf(qd_xyz, ligand_xyz, rtf_file=rtf_file)
>>> psf.write(psf_file)
Examples
If no ligand .xyz is on hand, or its atoms are in the wrong order, it is possible the extract the ligand directly from the quantum dot. This is demonstrated below with oleate (\(C_{18} H_{33} O_{2}^{-}\)).
>>> from scm.plams import Molecule
>>> from FOX import PSFContainer
>>> from FOX.recipes import generate_psf, extract_ligand
>>> qd = Molecule(...) # Accepts an .xyz, .pdb, .mol or .mol2 file
>>> rtf_file : str = ...
>>> ligand_len = 18 + 33 + 2
>>> ligand_atoms = {'C', 'H', 'O'}
>>> ligand: Molecule = extract_ligand(qd, ligand_len, ligand_atoms)
>>> psf: PSFContainer = generate_psf(qd, ligand, rtf_file=rtf_file)
>>> psf.write(...)
Examples
Example for multiple ligands.
>>> from typing import List
>>> from scm.plams import Molecule
>>> from FOX import PSFContainer
>>> from FOX.recipes import generate_psf2
>>> qd = Molecule(...) # Accepts an .xyz, .pdb, .mol or .mol2 file
>>> ligands = ('C[O-]', 'CC[O-]', 'CCC[O-]')
>>> rtf_files = (..., ..., ...)
>>> psf: PSFContainer = generate_psf2(qd, *ligands, rtf_file=rtf_files)
>>> psf.write(...)
If the the psf construction with generate_psf2() failes to identify a particular ligand,
it is possible to return all (failed) potential ligands with the ret_failed_lig parameter.
>>> ...
>>> ligands = ('CCCCCCCCC[O-]', 'CCCCBr')
>>> failed_mol_list: List[Molecule] = generate_psf2(qd, *ligands, ret_failed_lig=True)
Index¶
|
Generate a |
|
Generate a |
|
Extract a single ligand from qd. |
API¶
- FOX.recipes.generate_psf(qd, ligand=None, rtf_file=None, str_file=None)[source]¶
Generate a
PSFContainerinstance for qd.- Parameters:
qd (
strorMolecule) – The ligand-pacifated quantum dot. Should be supplied as either a Molecule or .xyz file.ligand (
strorMolecule, optional) – A single ligand. Should be supplied as either a Molecule or .xyz file.rtf_file (
str, optional) – The path+filename of the ligand’s .rtf file. Used for assigning atom types. Alternativelly, one can supply a .str file with the str_file argument.str_file (
str, optional) – The path+filename of the ligand’s .str file. Used for assigning atom types. Alternativelly, one can supply a .rtf file with the rtf_file argument.
- Returns:
A PSFContainer instance with the new .psf file.
- Return type:
PSFContainer
- FOX.recipes.generate_psf2(qd, *ligands, rtf_file=None, str_file=None, ret_failed_lig=False)[source]¶
Generate a
PSFContainerinstance for qd with multiple different ligands.Note
Requires the optional RDKit package.
- Parameters:
qd (
strorMolecule) – The ligand-pacifated quantum dot. Should be supplied as either a Molecule or .xyz file.*ligands (
str,MoleculeorChem.Mol) – One or more PLAMS/RDkit Molecules and/or SMILES strings representing ligands.rtf_file (
strorIterable[str], optional) – The path+filename of the ligand’s .rtf files. Filenames should be supplied in the same order as ligands. Used for assigning atom types. Alternativelly, one can supply a .str file with the str_file argument.str_file (
strorIterable[str], optional) – The path+filename of the ligand’s .str files. Filenames should be supplied in the same order as ligands. Used for assigning atom types. Alternativelly, one can supply a .rtf file with the rtf_file argument.ret_failed_lig (
bool) – IfTrue, return a list of all failed (potential) ligands if the function cannot identify any ligands within a certain range. Usefull for debugging. IfFalse, raise aMoleculeError.
- Returns:
A single ligand Molecule.
- Return type:
Molecule- Raises:
MoleculeError – Raised if the function fails to identify any ligands within a certain range. If
ret_failed_lig = True, return a list of failed (potential) ligands instead and issue a warning.
FOX.recipes.ligands¶
A set of functions for analyzing ligands.
Examples
An example for generating a ligand center of mass RDF.
>>> import numpy as np
>>> import pandas as pd
>>> from FOX import MultiMolecule, example_xyz
>>> from FOX.recipes import get_lig_center
>>> mol = MultiMolecule.from_xyz(example_xyz)
>>> start = 123 # Start of the ligands
>>> step = 4 # Size of the ligands
# Add dummy atoms to the ligand-center of mass and calculate the RDF
>>> lig_centra: np.ndarray = get_lig_center(mol, start, step)
>>> mol_new: MultiMolecule = mol.add_atoms(lig_centra, symbols='Xx')
>>> rdf: pd.DataFrame = mol_new.init_rdf(atom_subset=['Xx'])
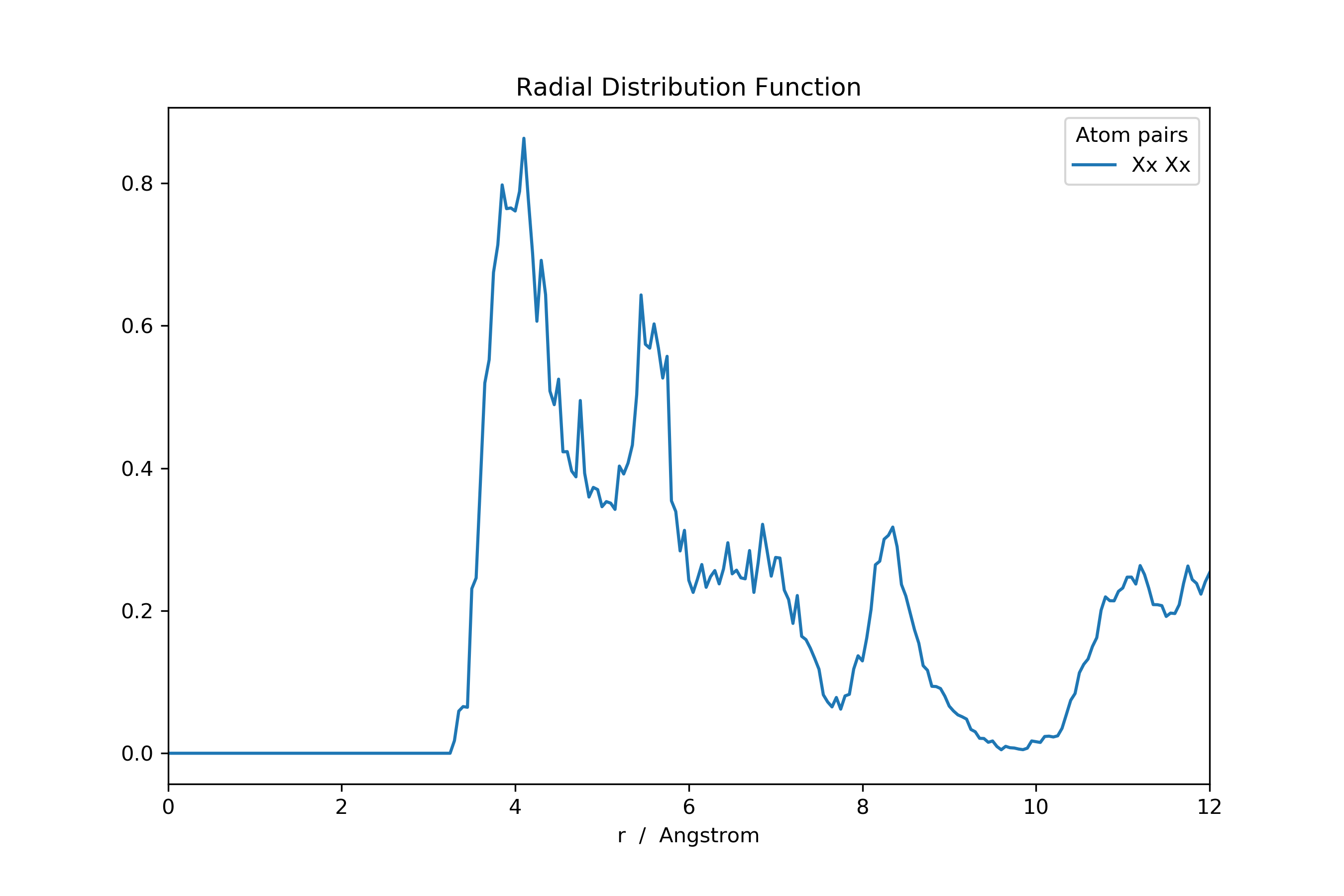
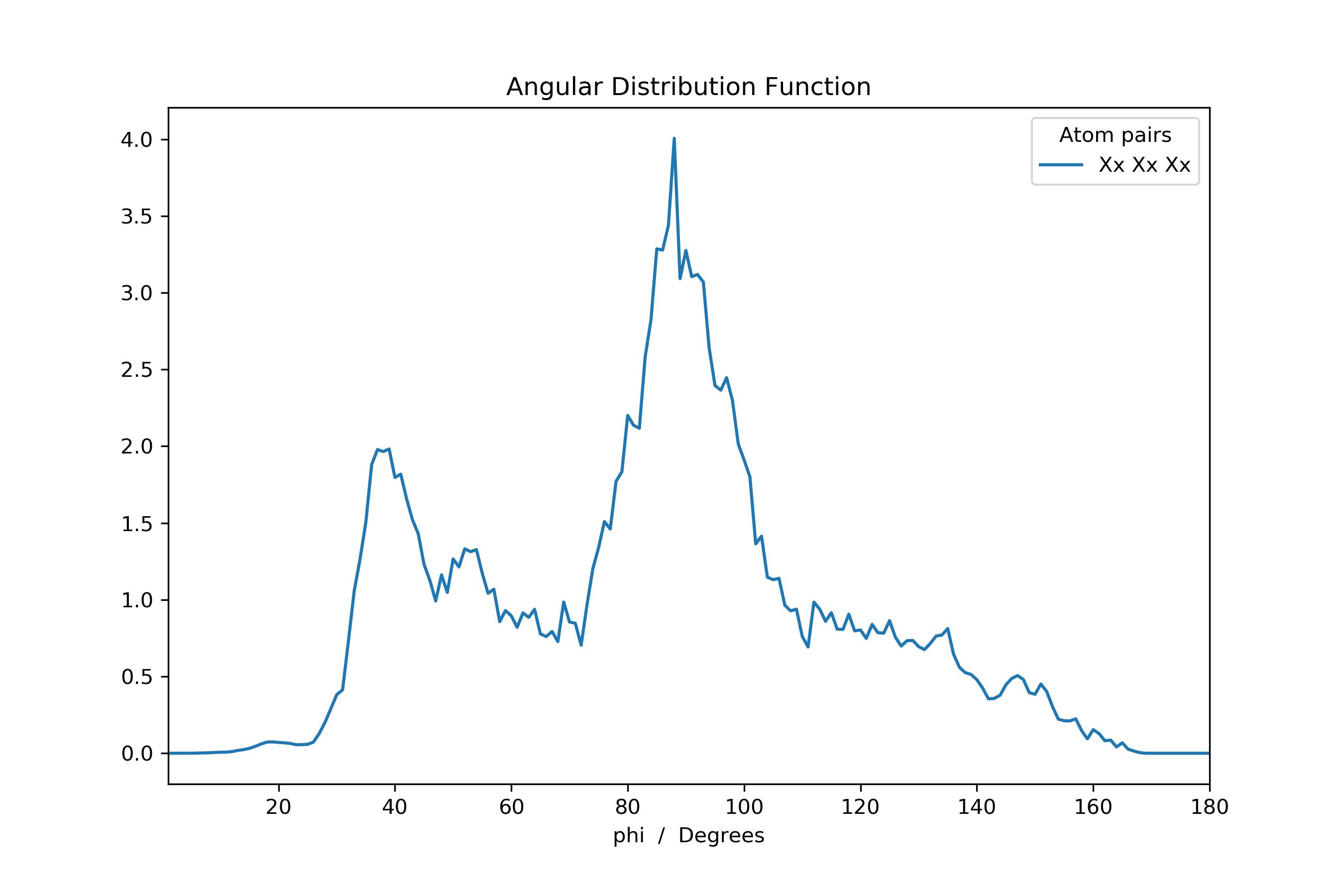
Or the ADF.
>>> ...
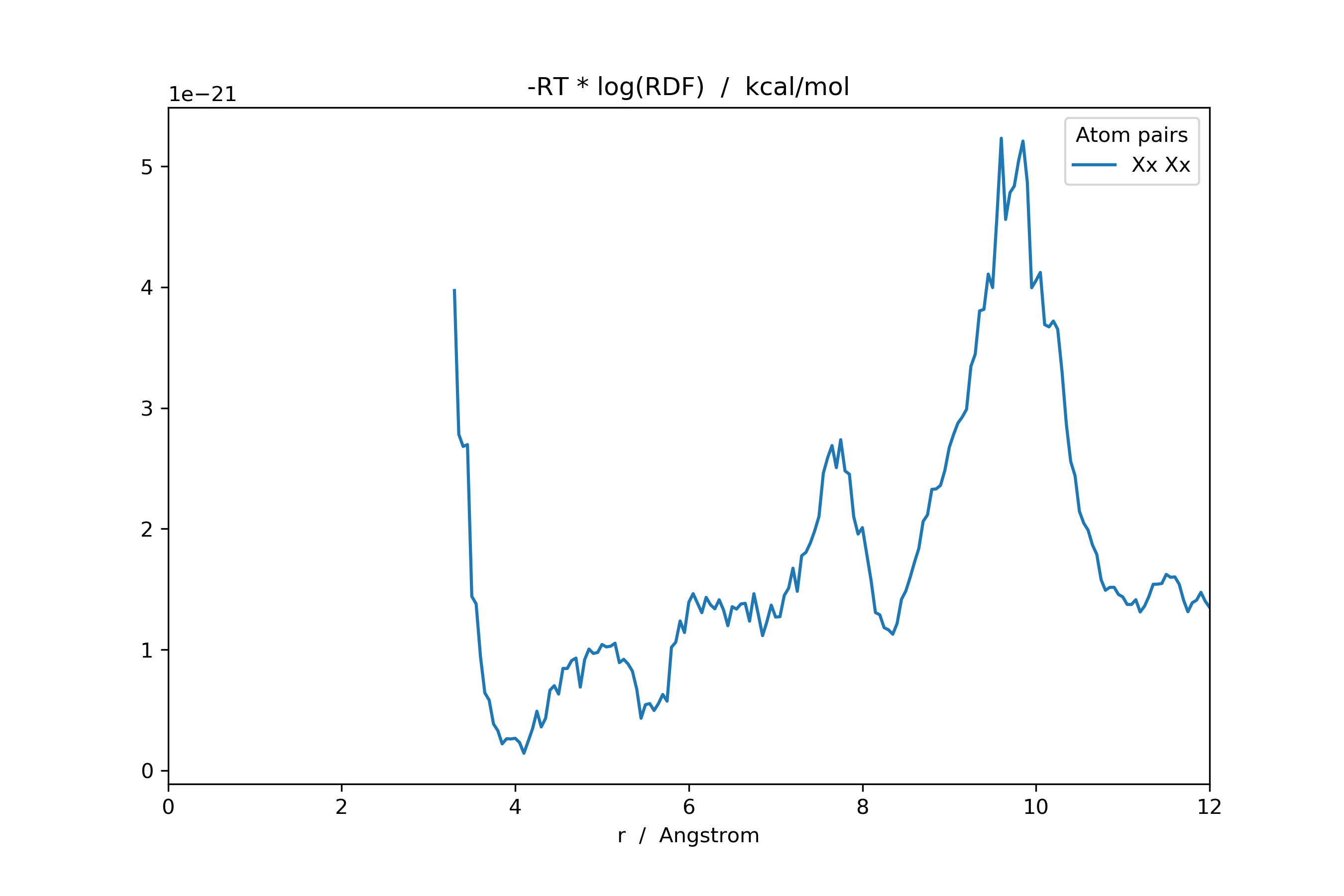
>>> adf: pd.DataFrame = mol_new.init_rdf(atom_subset=['Xx'], r_max=np.inf)
Or the potential of mean force (i.e. Boltzmann-inverted RDF).
>>> ...
>>> from scipy import constants
>>> from scm.plams import Units
>>> RT: float = 298.15 * constants.Boltzmann
>>> kj_to_kcal: float = Units.conversion_ratio('kj/mol', 'kcal/mol')
>>> with np.errstate(divide='ignore'):
>>> rdf_invert: pd.DataFrame = -RT * np.log(rdf) * kj_to_kcal
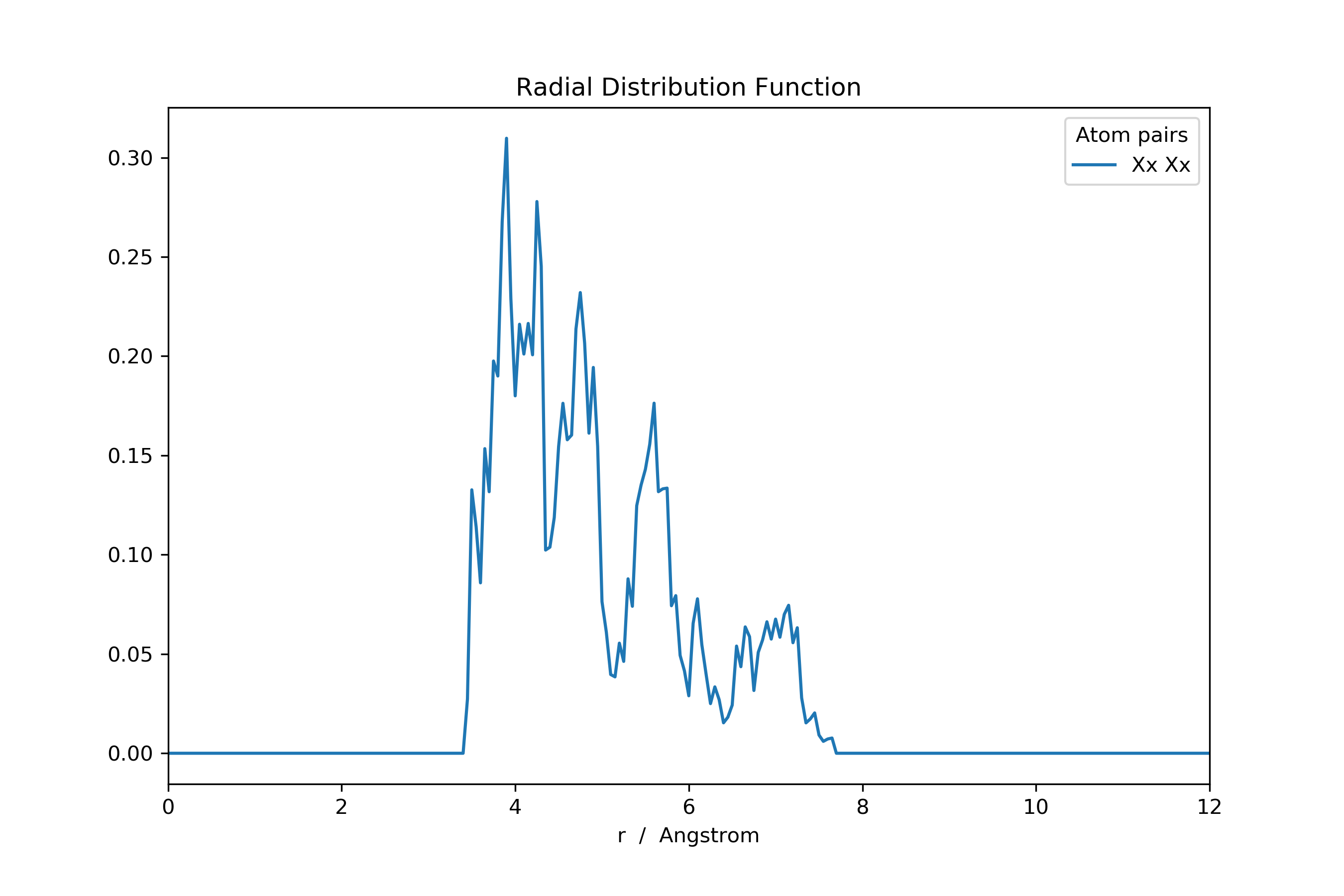
>>> rdf_invert[rdf_invert == np.inf] = np.nan # Set all infinities to not-a-number
Focus on a specific ligand subset is possible by slicing the new ligand Cartesian coordinate array.
>>> ...
>>> keep_lig = [0, 1, 2, 3] # Keep these ligands; disgard the rest
>>> lig_centra_subset = lig_centra[:, keep_lig]
# Add dummy atoms to the ligand-center of mass and calculate the RDF
>>> mol_new2: MultiMolecule = mol.add_atoms(lig_centra_subset, symbols='Xx')
>>> rdf: pd.DataFrame = mol_new2.init_rdf(atom_subset=['Xx'])
Examples
An example for generating a ligand center of mass RDF from a quantum dot with multiple unique ligands. A .psf file will herein be used as starting point.
>>> import numpy as np
>>> from FOX import PSFContainer, MultiMolecule, group_by_values
>>> from FOX.recipes import get_multi_lig_center
>>> mol = MultiMolecule.from_xyz(...)
>>> psf = PSFContainer.read(...)
# Gather the indices of each ligand
>>> idx_dict: dict = group_by_values(enumerate(psf.residue_id, start=1))
>>> del idx_dict[1] # Delete the core
# Use the .psf segment names as symbols
>>> symbols = [psf.segment_name[i].iloc[0] for i in idx_dict.values()]
# Add dummy atoms to the ligand-center of mass and calculate the RDF
>>> lig_centra: np.ndarray = get_multi_lig_center(mol, idx_dict.values())
>>> mol_new: MultiMolecule = mol.add_atoms(lig_centra, symbols=symbols)
>>> rdf = mol_new.init_rdf(atom_subset=set(symbols))
Index¶
|
Return an array with the (mass-weighted) mean position of each ligands in mol. |
|
Return an array with the (mass-weighted) mean position of each ligands in mol. |
API¶
- FOX.recipes.get_lig_center(mol, start, step, stop=None, mass_weighted=True)[source]¶
Return an array with the (mass-weighted) mean position of each ligands in mol.
- Parameters:
mol (
FOX.MultiMolecule) – A MultiMolecule instance.start (
int) – The atomic index of the first ligand atoms.step (
int) – The number of atoms per ligand.stop (
int, optional) – Can be used for neglecting any ligands beyond a user-specified atomic index.mass_weighted (
bool) – IfTrue, return the mass-weighted mean ligand position rather than its unweighted counterpart.
- Returns:
A new array with the ligand’s centra of mass. If
mol.shape == (m, n, 3)then, givenknew ligands, the to-be returned array’s shape is(m, k, 3).- Return type:
- FOX.recipes.get_multi_lig_center(mol, idx_iter, mass_weighted=True)[source]¶
Return an array with the (mass-weighted) mean position of each ligands in mol.
Contrary to
get_lig_center(), this function can handle molecules with multiple non-unique ligands.- Parameters:
mol (
FOX.MultiMolecule) – A MultiMolecule instance.idx_iter (
Iterable[Sequence[int]]) – An iterable consisting of integer sequences. Each integer sequence represents a single ligand (by its atomic indices).mass_weighted (
bool) – IfTrue, return the mass-weighted mean ligand position rather than its unweighted counterpart.
- Returns:
A new array with the ligand’s centra of mass. If
mol.shape == (m, n, 3)then, givenknew ligands (aka the length of idx_iter) , the to-be returned array’s shape is(m, k, 3).- Return type:
FOX.recipes.time_resolution¶
A set of functions for calculating time-resolved distribution functions.
Index¶
|
Calculate the time-resolved radial distribution function (RDF). |
|
Calculate the time-resolved angular distribution function (ADF). |
API¶
- FOX.recipes.time_resolved_rdf(mol, start=0, stop=None, step=500, **kwargs)[source]¶
Calculate the time-resolved radial distribution function (RDF).
Examples
>>> from FOX import MultiMolecule, example_xyz >>> from FOX.recipes import time_resolved_rdf # Calculate each RDF over the course of 500 frames >>> time_step = 500 >>> mol = MultiMolecule.from_xyz(example_xyz) >>> rdf_list = time_resolved_rdf( ... mol, step=time_step, atom_subset=['Cd', 'Se'] ... )
- Parameters:
mol (
MultiMolecule) – The trajectory in question.start (
int) – The initial frame.stop (
int, optional) – The final frame. Set toNoneto iterate over all frames.step (
int) – The number of frames per individual RDF. Note that lower step values will result in increased numerical noise.**kwargs (
Any) – Further keyword arguments forinit_rdf().
- Returns:
A list of dataframes, each containing an RDF calculated over the course of step frames.
- Return type:
See also
init_rdf()Calculate the radial distribution function.
- FOX.recipes.time_resolved_adf(mol, start=0, stop=None, step=500, **kwargs)[source]¶
Calculate the time-resolved angular distribution function (ADF).
Examples
>>> from FOX import MultiMolecule, example_xyz >>> from FOX.recipes import time_resolved_adf # Calculate each ADF over the course of 500 frames >>> time_step = 500 >>> mol = MultiMolecule.from_xyz(example_xyz) >>> rdf_list = time_resolved_adf( ... mol, step=time_step, atom_subset=['Cd', 'Se'] ... )
- Parameters:
mol (
MultiMolecule) – The trajectory in question.start (
int) – The initial frame.stop (
int, optional) – The final frame. Set toNoneto iterate over all frames.step (
int) – The number of frames per individual RDF. Note that lower step values will result in increased numerical noise.**kwargs (
Any) – Further keyword arguments forinit_adf().
- Returns:
A list of dataframes, each containing an ADF calculated over the course of step frames.
- Return type:
See also
init_adf()Calculate the angular distribution function.
FOX.recipes.similarity¶
Recipes for computing the similarity between trajectories.
Examples
An example where, starting from two .xyz files, the similarity is computed between two molecular dynamics (MD) trajectories.
>>> import numpy as np
>>> import FOX
>>> from FOX.recipes import compare_trajectories
# The relevant multi-xyz files
>>> md_filename: str = ...
>>> md = FOX.MultiMolecule.from_xyz(md_filename)
>>> md_ref_filename: str = ...
>>> md_ref = FOX.MultiMolecule.from_xyz(md_ref_filename)
# Calculate the similarity between `md` and `md_ref`
>>> similarity = compare_trajectories(md, md_ref, metric="cosine")
# Identify all sufficiently dissimilar molecules (as defined via `threshold`)
>>> threshold: float = ...
>>> idx = np.zeros(len(md), dtype=np.bool_)
>>> idx[similarity >= threshold] = True
The resulting indices can be used for, for example, identifying all molecules one wants to use for further (quantum-mechanical/classical) calculations.
>>> import qmflows
# Define the job settings
>>> s = qmflows.Settings()
>>> s.lattice = [50, 50, 50]
>>> s.specific.cp2k.motion.print["forces on"].filename = ""
>>> s.overlay(qmflows.templates.singlepoint)
# Construct the job list
>>> mol_list = md[idx].as_Molecule()
>>> job_list = [qmflows.cp2k(s, mol) for mol in mol_list]
# Run the jobs
>>> result_list = [qmflows.run(job) for job in job_list]
# Extract the forces and energies from all jobs
>>> forces = np.array([r.forces for r in result_list])[:, 0]
>>> energy = np.array([r.energy for r in result_list])[:, 0]
Index¶
|
Compute the similarity between 2 trajectories according to the specified metric. |
|
Return the indices that yield a uniform distribution of n points. |
API¶
- FOX.recipes.compare_trajectories(md, md_ref, *, metric='cosine', reduce=<function mean>, reset_origin=True, **kwargs)[source]¶
Compute the similarity between 2 trajectories according to the specified metric.
The default metric aliases
scipy.spatial.distance.cdist()for defining the (dis-)similarity between the passed md and its reference. This (dis-)similarity array is subsequently reduced to a vector of size \((N_{mol},)\) by taking its mean (along the relevant axes).Examples
>>> import numpy as np >>> from FOX.recipes import compare_trajectories >>> md: np.ndarray = ... >>> md_ref: np.ndarray = ... # Default `metric` presets >>> metric1 = compare_trajectories(md, md_ref, metric="cosine") >>> metric2 = compare_trajectories(md, md_ref, metric="euclidean") >>> metric3 = compare_trajectories(md, md_ref, metric="minkowski", p=1) >>> def rmsd(a: np.ndarray) -> np.float64: ... '''Calculate the root-mean-square deviation.''' ... return np.mean(a**2)**0.5 # Sum over the number of atoms rather than average >>> metric4 = compare_trajectories(md, md_ref, reduce=np.sum) >>> metric5 = compare_trajectories(md, md_ref, reduce=rmsd) >>> def sqeuclidean(md: np.ndarray, md_ref: np.ndarray) -> np.ndarray: ... '''Calculate the distance based on the squared eclidian norm.''' ... delta = md[..., None] - md_ref[..., None, :] ... return np.linalg.norm(delta, axis=-1)**2 # Pass a custom metric-function >>> metric6 = compare_trajectories(md, md_ref, metric=sqeuclidean)
- Parameters:
md (array_like, shape \((N_{mol}, N_{atom1}, 3)\) or \((N_{atom1}, 3)\)) – An array-like object containing the trajectory of interest.
md_ref (array_like, shape \((N_{mol}, N_{atom2}, 3)\) or \((N_{atom2}, 3)\)) – An array-like object containing the reference trajectory.
metric (
strorCallable[[FOX.MultiMolecule, FOX.MultiMolecule], np.ndarray]) – The type of metric used for calculating the (dis-)similarity. Accepts either a callback or predefined alias. See metric parameter inscipy.spatial.distance.cdist()for a comprehensive overview of all aliases. If a callback is provided then it should take a array of shape \((n_{atom1}, 3)\) and \((N_{atom2}, 3)\) as arguments and return a new array of shape \((N_{atom1}, N_{atom2})\).reduce (
Callable[[np.ndarray], np.number], optional) – A callable for performing a dimensional reduction. Used for transforming the shape \((N_{atom1}, N_{atom2})\) array, returned by metric, into a scalar. Setting this value toNonewill disable the reduction and return the metric output in unaltered form.reset_origin (
bool) – Reset the origin by removing translations and rotations from the passed trajectories.**kwargs (
Any) – Further keyword arguments for metric.
- Returns:
An array with the (dis-)similarity between all molecules in md and md_ref.
- Return type:
np.ndarray[np.float64], shape \((N_{mol},)\)
See also
scipy.spatial.distance.cdist()Compute distance between each pair of the two collections of inputs.
- FOX.recipes.fps_reduce(dist_mat, n=1, **kwargs)[source]¶
Return the indices that yield a uniform distribution of n points.
Examples
>>> from functools import partial >>> import numpy as np >>> from FOX.recipes import compare_trajectories, fps_reduce >>> md: np.ndarray = ... >>> md_ref: np.ndarray = ... >>> reduce_func = partial(fps_reduce, n=10) >>> out = compare_trajectories(md, md_ref, reduce=reduce_func)
Note
This function requires the Compound Attachment Tools package: CAT.
- Parameters:
dist_mat (
np.ndarray[np.float64], shape \((m_a, m_b)\)) – A distance matrix.n (
int, optional) – The number of to-be returned indices.**kwargs (
Any) – Further keyword arguments forCAT.distribution.uniform_idx().
- Returns:
An array of indices.
- Return type:
np.ndarray[np.int64], shape \((n,)\)
See also
CAT.distribution.uniform_idx()Yield the column-indices that result in a uniform or clustered distribution.
FOX.recipes.compare_trajectories()Compute the similarity between 2 trajectories according to the specified metric.
FOX.recipes.top¶
Recipe for creating GROMACS .top files from an .xyz and CHARMM .rtf and .str files.
Index¶
|
Construct a |
API¶
- FOX.recipes.create_top(*, mol_count, rtf_files, prm_files, generate_14_nb_pairs=True, generate_nb_pairs=True)[source]¶
Construct a
FOX.TOPContainerobject from the passed CHARMM .rtf and .prm files.Examples
>>> from FOX.recipes import create_top >>> output_path: str = ... >>> rtf_files = ["ligand1.rtf", "ligand2.rtf"] >>> prm_files = ["ligand1.prm", "ligand2.prm"] >>> mol_count = [30, 15] # 30 ligand1 residues and 15 ligand2 residues >>> top = create_top( ... mol_count=mol_count, rtf_files=rtf_files, prm_files=prm_files, ... ) >>> top.to_file(output_path)
- Parameters:
mol_count (
list[int]) – The number of molecules of a given residue. Note that rtf files may contain multiple residues.rtf_files (list of path-like objects) – The names of all to-be converted .rtf files
prm_files (list of path-like and/or
FOX.PRMContainerobjects) – The names of all to-be converted .prm filesgenerate_14_nb_pairs (bool) – Whether to automatically generate all 1,4 non-bonded pairs
generate_nb_pairs (bool) – Whether to automatically generate non-bonded pairs for all (indirectly) unconnected atoms.
- Returns:
A new .top container object
- Return type:
FOX.TOPContainer
FOX.recipes.xyz_to_gro¶
Interconvert between .xyz and .gro files.
Examples
This recipe is available from the command line via the FOX.recipes.xyz_to_gro entry point:
> FOX.recipes.xyz_to_gro file.xyz file.gro
Index¶
|
Convert the passed .xyz file into a .gro file. |
|
Convert the passed .xyz file into a .gro file. |