Root Mean Squared Displacement & Fluctuation¶
Root Mean Squared Displacement¶
The root mean squared displacement (RMSD) represents the average displacement of a set or subset of atoms as a function of time or, equivalently, moleculair indices in a MD trajectory.
Given a trajectory, mol, stored as a FOX.MultiMolecule instance,
the RMSD can be calculated with the FOX.MultiMolecule.init_rmsd()
method using the following command:
>>> rmsd = mol.init_rmsd(atom_subset=None)
The resulting rmsd is a Pandas dataframe, an object which is effectively a
hybrid between a dictionary and a NumPy array.
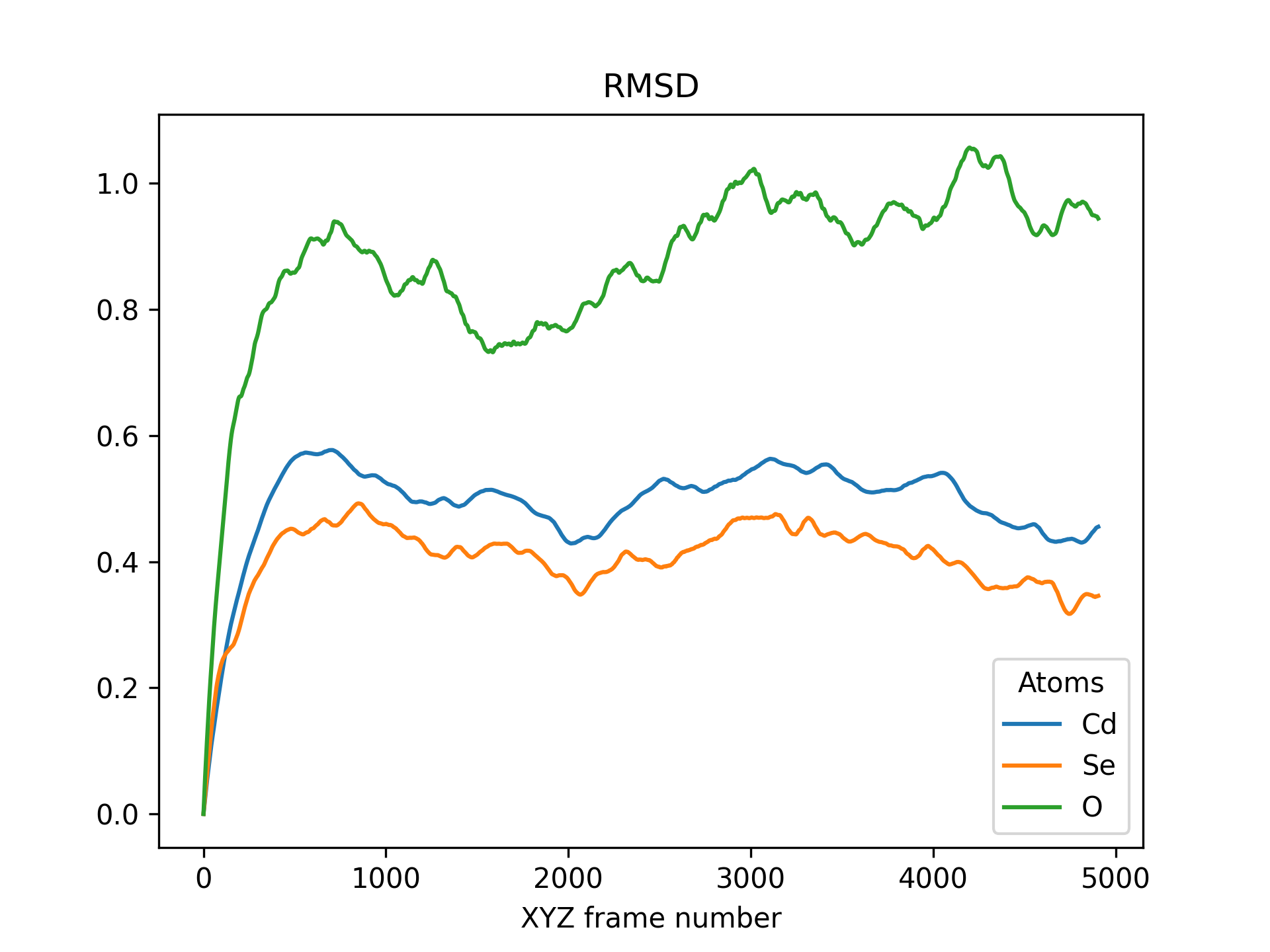
Below is an example RMSD of a CdSe quantum dot pacified with formate ligands. The RMSD is printed for cadmium, selenium and oxygen atoms.
>>> from FOX import MultiMolecule, example_xyz
>>> mol = MultiMolecule.from_xyz(example_xyz)
>>> rmsd = mol.init_rmsd(atom_subset=('Cd', 'Se', 'O'))
>>> rmsd.plot(title='RMSD')
Root Mean Squared Fluctuation¶
The root mean squared fluctuation (RMSD) represents the time-averaged displacement, with respect to the time-averaged position, as a function of atomic indices.
Given a trajectory, mol, stored as a FOX.MultiMolecule instance,
the RMSF can be calculated with the FOX.MultiMolecule.init_rmsf()
method using the following command:
>>> rmsd = mol.init_rmsf(atom_subset=None)
The resulting rmsf is a Pandas dataframe, an object which is effectively a
hybrid between a dictionary and a Numpy array.
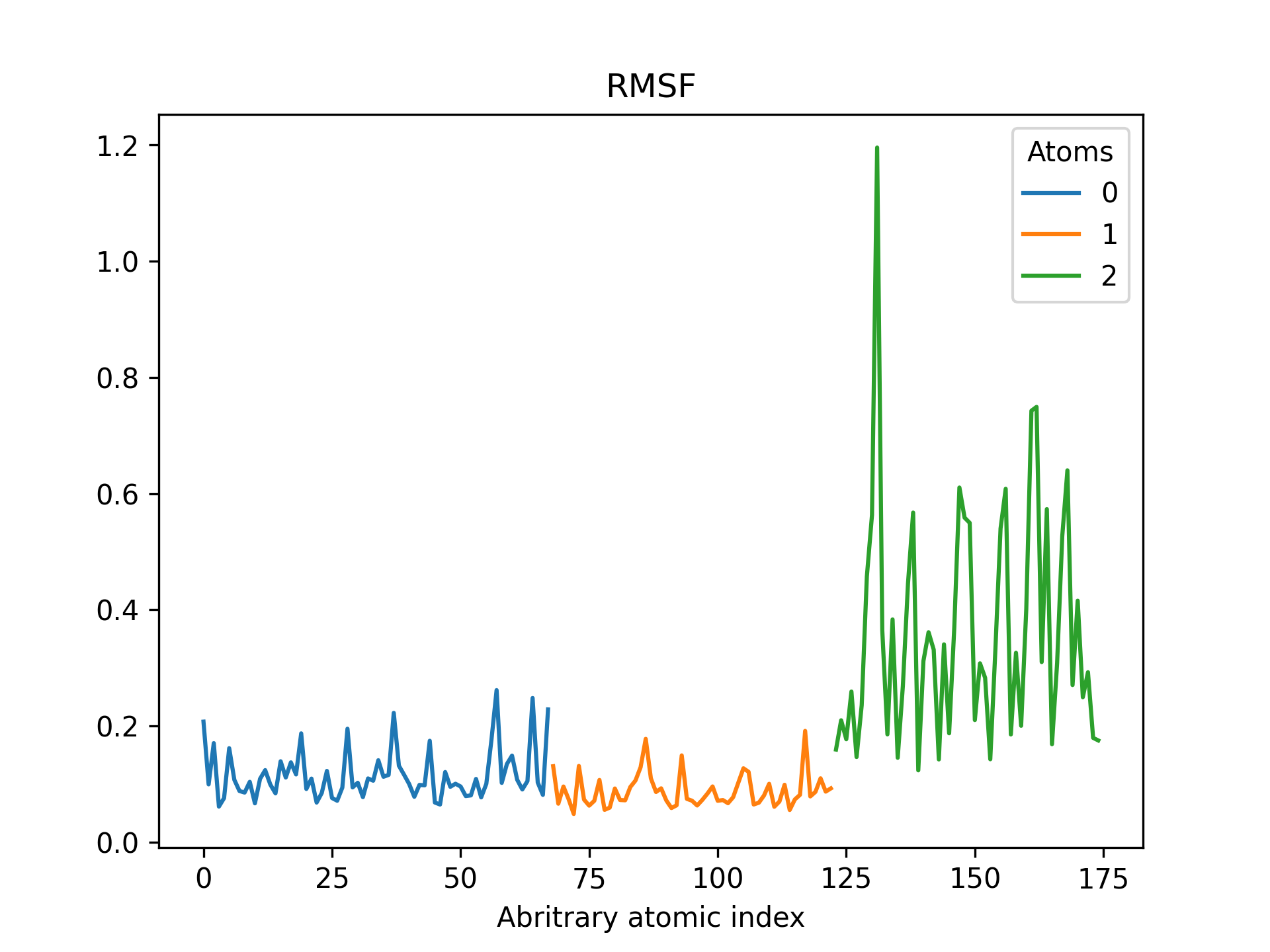
Below is an example RMSF of a CdSe quantum dot pacified with formate ligands. The RMSF is printed for cadmium, selenium and oxygen atoms.
>>> from FOX import MultiMolecule, example_xyz
>>> mol = MultiMolecule.from_xyz(example_xyz)
>>> rmsd = mol.init_rmsf(atom_subset=('Cd', 'Se', 'O'))
>>> rmsd.plot(title='RMSF')
The atom_subset argument¶
In the above two examples atom_subset=None was used an optional keyword,
one which allows one to customize for which atoms the RMSD & RMSF should be
calculated and how the results are distributed over the various columns.
There are a total of four different approaches to the atom_subset argument:
1. atom_subset=None: Examine all atoms and store the results in a single column.
2. atom_subset=int: Examine a single atom, based on its index, and store the results in a single column.
3. atom_subset=str or atom_subset=list(int): Examine multiple atoms, based on their atom type or indices, and store the results in a single column.
4. atom_subset=tuple(str) or atom_subset=tuple(list(int)): Examine multiple atoms, based on their atom types or indices, and store the results in multiple columns. A column is created for each string or nested list in atoms.
It should be noted that lists and/or tuples can be interchanged for any other iterable container (e.g. a Numpy array), as long as the iterables elements can be accessed by their index.
API¶
- MultiMolecule.init_rmsd(mol_subset=None, atom_subset=None, reset_origin=True)[source]
Initialize the RMSD calculation, returning a dataframe.
- Parameters:
mol_subset (
slice, optional) – Perform the calculation on a subset of molecules in this instance, as determined by their moleculair index. Include all \(m\) molecules in this instance ifNone.atom_subset (
Sequence[str], optional) – Perform the calculation on a subset of atoms in this instance, as determined by their atomic index or atomic symbol. Include all \(n\) atoms per molecule in this instance ifNone.reset_origin (
bool) – Reset the origin of each molecule in this instance by means of a partial Procrustes superimposition, translating and rotating the molecules.
- Returns:
A dataframe of RMSDs with one column for every string or list of ints in atom_subset. Keys consist of atomic symbols (e.g.
"Cd") if atom_subset contains strings, otherwise a more generic ‘series ‘ + str(int) scheme is adopted (e.g."series 2"). Molecular indices are used as index.- Return type:
- MultiMolecule.init_rmsf(mol_subset=None, atom_subset=None, reset_origin=True)[source]
Initialize the RMSF calculation, returning a dataframe.
- Parameters:
mol_subset (
slice, optional) – Perform the calculation on a subset of molecules in this instance, as determined by their moleculair index. Include all \(m\) molecules in this instance ifNone.atom_subset (
Sequence[str], optional) – Perform the calculation on a subset of atoms in this instance, as determined by their atomic index or atomic symbol. Include all \(n\) atoms per molecule in this instance ifNone.reset_origin (
bool) – Reset the origin of each molecule in this instance by means of a partial Procrustes superimposition, translating and rotating the molecules.
- Returns:
A dataframe of RMSFs with one column for every string or list of ints in atom_subset. Keys consist of atomic symbols (e.g.
"Cd") if atom_subset contains strings, otherwise a more generic ‘series ‘ + str(int) scheme is adopted (e.g."series 2"). Molecular indices are used as indices.- Return type:
- MultiMolecule.init_shell_search(mol_subset=None, atom_subset=None, rdf_cutoff=0.5)[source]
Calculate and return properties which can help determining shell structures.
Warning
Depercated.
- static MultiMolecule.get_at_idx(rmsf, idx_series, dist_dict)[source]
Create subsets of atomic indices.
Warning
Depercated.